As terapêuticas clínicas da hipertensão arterial pulmonar
Redação
A hipertensão pulmonar é uma síndrome clínica e hemodinâmica que resulta no aumento da resistência vascular na pequena circulação, elevando os níveis pressóricos na circulação pulmonar. Este aumento de resistência pode estar associado a várias condições médicas subjacentes ou a uma doença que afete exclusivamente a circulação pulmonar.
O tratamento inicial inclui os bloqueadores dos Canais de Cálcio (BCC) e a nifedipino ou anlodipino são utilizados em pacientes com teste de vasorreatividade pulmonar positivo. A dose deve ser a maior tolerável, e a resposta deve ser avaliada após 3 a 6 meses.
Para as terapias específicas, os inibidores da fosfodiesterase 5 (PDE5i), como a sildenafila é uma opção para melhorar a capacidade de exercício e reduzir a pressão arterial pulmonar. Os antagonistas de receptor da endotelina (ERA), como a ambrisentana e bosentana são utilizados para melhorar a capacidade física e diminuir a taxa de agravamento clínico.
Para pacientes que não alcançam parâmetros de baixo risco com monoterapia, pode-se considerar a terapia combinada, que pode incluir uma combinação de BCC, PDE5i, ERA e prostanoides. Como tratamento adjuvante, os anticoagulantes são considerados em pacientes com HAP idiopática e fatores de risco para tromboembolismo.
Os diuréticos são utilizados em casos de descompensação de insuficiência cardíaca direita e retenção hídrica. A resposta ao tratamento deve ser monitorada regularmente, e ajustes na terapia devem ser feitos conforme necessário, especialmente se o paciente não apresentar melhora ou se a condição piorar.
A escolha do tratamento deve levar em conta a classe funcional do paciente, a presença de comorbidades e a resposta ao tratamento inicial. A terapia deve ser individualizada e pode incluir intervenções não farmacológicas, como suporte psicológico e restrição de sódio na dieta.
Conforme o MS-PCDT: Hipertensão Pulmonar, apesar de ser considerada uma doença rara, a HP está se tornando um problema de saúde global cada vez mais comum e associada a um prognóstico ruim. Dados epidemiológicos estimam que a incidência mundial de HP seja entre 2 e 5 pacientes acometidos a cada milhão de adultos por ano, sendo que a incidência aumenta em indivíduos com idade acima de 65 anos.
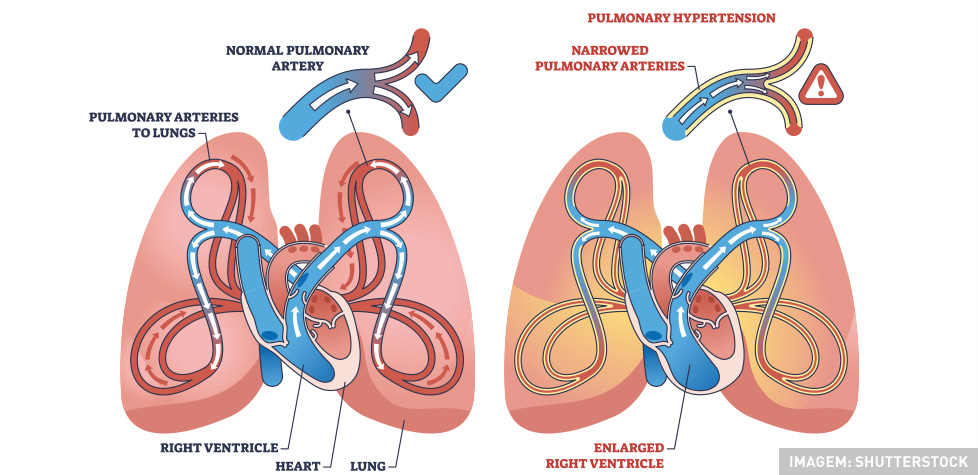
Uma alta carga de mortalidade está associada à doença, principalmente quando há ausência de tratamento específico, casos nos quais é estimada uma sobrevida mediana de 2,8 anos. A definição de HP já foi bastante discutida e ajustada ao longo dos anos. A definição atual, elaborada por consenso por ocasião do 6º Simpósio Mundial de HP, estabelece que a HP pode ser definida por pressão arterial pulmonar média (PAPm) acima de 20 mmHg combinada a outras medidas hemodinâmicas, como, por exemplo, pressão de oclusão capilar pulmonar (POCP) e resistência vascular pulmonar (RVP).
Conforme a variação das medidas hemodinâmicas, a HP pode ser classificada em pré-capilar, em pós-capilar e na combinação de pré e pós-capilar, como apresentado no Quadro 1. A HP pré-capilar é definida pela presença concomitante de PAPm maior do que 20 mmHg, POCP menor ou igual a 15 mmHg e RVP igual ou maior a 3 unidades Wood.
Nesta situação, o predomínio da doença vascular está no território arterial e há a necessidade de cateterismo cardíaco direito, como medida mandatória do débito cardíaco (DC) e da POCP. A falha na confirmação do diagnóstico por cateterismo cardíaco direito é a principal causa de excesso de diagnóstico e tratamento da HP pré-capilar.
Por outro lado, quando a POCP for superior a 15 mmHg, o paciente apresenta HP pós-capilar, o que sugere a presença de alterações nas câmaras cardíacas esquerdas. Há situações em que a HP pode ser classificada como combinada, isto é, quando a POCP está acima de 15 mmHg, mas não parece suficiente para justificar a magnitude de elevação da PAPm.
Esses pacientes apresentam resistência vascular pulmonar maior ou igual a 3 unidades Wood e geralmente o gradiente diastólico pulmonar superior a 7 mmHg (GDP - diferença entre a pressão diastólica de artéria pulmonar e a pressão de capilar pulmonar). Os sinais e sintomas de HP são bastante semelhantes aos de outras causas de insuficiência respiratória crônica, como dispneia progressiva, fadiga crônica, fraqueza, angina, estase jugular, cianose, pré-síncope e síncope.
Os achados físicos podem incluir elevação ou retração paraesternal esquerda, segunda bulha cardíaca aumentada, terceira bulha cardíaca do ventrículo direito, pressão venosa jugular elevada com forma de onda anormal, pulsos arteriais de baixo volume, hepatomegalia, ascite, edema periférico e sopro regurgitante tricúspide. Como várias doenças apresentam acometimento vascular pulmonar frequente, o médico deve estar atento para a possibilidade de HP, em especial a esclerodermia (até 27% de prevalência de HP), esquistossomose (7,7% de HP na doença hepatoesplênica), hipertensão portal (7,2% de HP em candidatos à transplante hepático), infecção pelo HIV (0,5% de HP) e embolia pulmonar (5,1% de prevalência de HPTEC).
Artigo atualizado em 11/11/2025 11:35.