As diretrizes terapêuticas da fibrose cística
Redação
A fibrose cística ou mucoviscidose é uma doença genética e hereditária que afeta principalmente os pulmões e o sistema digestivo, causando acúmulo de muco espesso e pegajoso. Essa condição pode levar a problemas respiratórios crônicos, infecções frequentes e dificuldades na absorção de nutrientes.
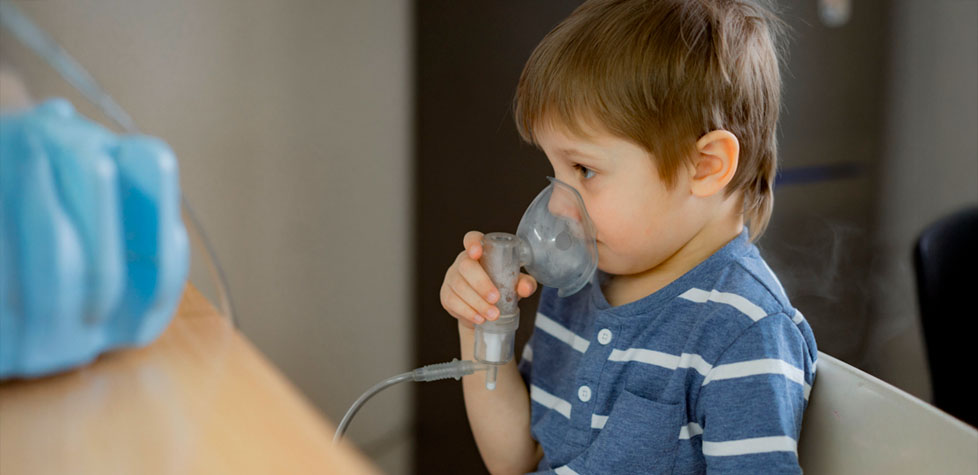
A fibrose cística ou mucoviscidose é uma doença genética e hereditária que afeta principalmente os pulmões e o sistema digestivo, causando acúmulo de muco espesso e pegajoso. Essa condição pode levar a problemas respiratórios crônicos, infecções frequentes e dificuldades na absorção de nutrientes.
As diretrizes terapêuticas estabelecem o diagnóstico, tratamento e acompanhamento dos indivíduos com essa condição. Assim, o diagnóstico deve ser realizado com base na triagem neonatal ou suspeita clínica, utilizando o teste do suor e genotipagem do gene CFTR. O tratamento deve ser realizado em centros de referência por uma equipe multidisciplinar, incluindo pneumologistas, gastroenterologistas, fisioterapeutas, nutricionistas, enfermeiros, psicólogos, assistentes sociais e farmacêuticos.
A terapia medicamentosa pode incluir moduladores da proteína CFTR, como ivacaftor e elexacaftor/tezacaftor/ivacaftor, dependendo das mutações genéticas dos pacientes. Os pacientes devem ser avaliados periodicamente quanto à eficácia do tratamento e ao desenvolvimento de toxicidade.
A adesão ao tratamento deve ser monitorada, e intervenções psicossociais podem ser necessárias para melhorar a adesão. Os pacientes com diagnóstico de fibrose cística e certas características clínicas são elegíveis para tratamentos específicos, enquanto aqueles com bronquiectasias ou insuficiência pancreática de outras etiologias devem ser excluídos.
O tratamento visa promover crescimento e desenvolvimento saudável, melhorar a função pulmonar, aumentar a sobrevida e reduzir complicações. Essas diretrizes são fundamentais para garantir que os pacientes recebam um tratamento adequado e eficaz, melhorando sua qualidade de vida e prognóstico.
O MS-PCDT: Fibrose Cística de 2025 detalha que a fibrose cística (FC) é uma doença genética com acometimento multissistêmico, decorrente de variantes patogênicas em ambos os alelos do gene Regulador de Condutância Transmembrana (Cystic Fibrosis Transmembrane Conductance Regulator – CFTR), codificador da proteína CFTR, um canal de cloreto e bicarbonato presente na superfície apical das células epiteliais do organismo e que bombeia substratos de forma ativa através das membranas.
Existem mais de 2 mil mutações identificadas no gene CFTR registradas na base de dados Cystic Fibrosis Mutation Database. Essas mutações são classificadas em seis classes distintas, conforme o tipo de defeito que causam na proteína CFTR, de maior ou menor expressão ou alteração de sua função nas células epiteliais: - Classe I (produção): ausência da proteína ou proteína truncada, levando à perda completa ou quase completa da função da proteína CFTR; - Classe II (processamento): síntese de uma proteína imatura, com pouca ou nenhuma proteína na membrana apical. Nesta classe, a mutação mais frequente é a Phe508del; - Classe III (regulação): a regulação é defeituosa e a proteína não pode ser ativada, apesar de haver expressão de CFTR; - Classe IV (condução): a condutância do cloreto é diminuída, apesar de haver síntese e expressão da CFTR, com função residual da proteína na membrana; pode levar a fenótipo de menor gravidade; - Classe V (síntese reduzida): síntese da CFTR parcialmente prejudicada, com quantidade reduzida.
Podem levar a fenótipo de menor gravidade; e - Classe VI (degradação acelerada): proteína com instabilidade na membrana apical da célula, com degradação 5 a 6 vezes mais veloz do que a observada com a proteína selvagem.
Estima-se que existe, em todo o mundo, mais de 105 mil doentes com FC e, no Brasil, o rastreamento obrigatório da FC pelo teste de triagem neonatal, com dosagem do tripsinogênio imunorreativo, tem aumentado o diagnóstico, com diminuição da mediana da idade de diagnóstico para menos de 3 meses, e elevado o conhecimento sobre o perfil genético dos pacientes. Anualmente, são diagnosticados em torno de 300 casos e mais de 6,4 mil pacientes estão cadastrados no Registro Brasileiro de Fibrose Cística (REBRAFC), dos quais 69% residem nas regiões Sudeste e Sul do Brasil.
A incidência da doença na população brasileira é amplamente variável, conforme a região geográfica e o grau de miscigenação populacional, variando entre 1:1.000 nos estados da região Sul até 1:10.000 em São Paulo. Além disso, dados de 2021 mostraram que a maior parte das pessoas com a doença são crianças e adolescentes, com uma média de idade de 13,43 anos, sendo que 25,64% do total dos casos é de pessoas com idade igual ou superior a 18 anos.
Os avanços nesse campo de conhecimento, a possibilidade de diagnóstico precoce e as melhorias no cuidado têm aumentado a expectativa de vida dos pacientes. Em alguns países, a população pediátrica corresponde a menos da metade das pessoas com FC, cuja idade mediana de morte é de 50 anos. Contudo, a FC está associada a morbidade significativa e elevada mortalidade.
Em sua forma grave, as manifestações multissistêmicas são observadas, incluindo doença pulmonar, insuficiência pancreática exócrina e outras manifestações gastrointestinais, sinusite crônica, diabetes, entre outros sintomas. Já em quadros mais leves, as manifestações são mais tardias ou observadas em apenas um sistema, tal como a obstrução de ducto deferente em homens com infertilidade, sendo muitas vezes referido como doença relacionada ao CFTR, má absorção de nutrientes, risco aumentado de desidratação e distúrbios metabólicos.
Artigo atualizado em 25/08/2025 03:00.